Mucopolysaccharidosis Type II in Zambia: A case report highlighting the importance of multi-level collaborations in rare diseases globally
Derby Tembo
Chipata Central Hospital, Dept. of Paediatrics and Child Health, Chipata, Zambia
University Teaching Hospitals, Dept. of Paediatrics and Child Health, Lusaka, Zambia
University Teaching Hospitals Neurology Research Office, Lusaka, Zambia.
Swedy Nkhoma
Solwezi General Hospital, Dept. of Paediatrics and Child Health, Kitwe, Zambia.
Kafula Lisa Nkole
University Teaching Hospitals, Dept. of Paediatrics and Child Health, Lusaka, Zambia
Nfwama Kawatu
University Teaching Hospitals, Dept. of Paediatrics and Child Health, Lusaka, Zambia
Archana A. Patel
Boston Children’s Hospital, Dept of Neurology, Division of Epilepsy and Clinical neurophysiology.
University of Zambia School of Medicine, Dept. of Paediatrics and Child Health, Lusaka, Zambia
DOI: https://doi.org/10.55320/mjz.51.2.543
Keywords:Iduronate-2-sulphatase, glycosaminoglycan, X-linked, Hunter syndrome, lysosomal storage disorder, multidisciplinary care.
ABSTRACT
Mucopolysaccharidosis type II (MPS II) is an X-linked lysosomal storage disorder with an insidious onset and a non-reversible progressive course caused by a mutation in a gene that codes for iduronate-2-sulfatase (IDS), a lysosomal enzyme leading to a systemic accumulation of glycosaminoglycan (GAGs). Definitive diagnosis is made by the demonstration of heparan and dermatan sulfatase in urine, enzyme activity assay and molecular analysis of the IDS gene. Management primarily focuses on symptomatic treatment via a multidisciplinary team approach; and where accessible, enzyme replacement therapy. Early diagnosis and opportune commencement of therapy are important to improve quality of life, reduce morbidity and increased expectancy of life. As confirmatory diagnostic testing is limited in many low resource settings, institutional collaborations is key to diagnosing rare diseases. We describe the clinical course, diagnosis and management of an 11-year-old boy from rural Zambia.
INTRODUCTION
Mucopolysaccharidosis type II (MPS II; also known as Hunter syndrome) is a multi-systemic X-linked recessive lysosomal storage disorder caused by a mutation in the iduronate-2-sulfatase (IDS) gene, an enzyme involved in the catabolism of glycosaminoglycan (GAGs). This leads to accumulation of dermatan and heparan sulphate.[1,2]
The global incidence and prevalence of MPS II is estimated to be 0.3–0.71 per 100,000 live births and 1.3:100 000 male live births respectively.[2,3] Despite the disease affecting mostly males due to its X-linked inheritance, some rare cases in females with associated structural abnormalities, X chromosome inactivation and X chromosome monosomy have been reported.[4,5,6] There is limited data on the incidence of MPS II in Africa, with only a few cases reported in various parts of the region.[7,8,9,10] In Zambia, the only published report of MPS II to date was descriptive, due to lack of confirmatory diagnostic capacity at time of report.[11]
The onset of symptoms is insidious caused by a non-reversible progressive accumulation of the GAGs in the intracellular and extracellular spaces. The phenotype varies greatly from mild to severe forms and should be regarded as a continuum between the two extremes. Absolute enzyme activity is not predictive of the severity of phenotype, suggesting that other factors might be implicated.[3] Milder forms commonly manifest between the ages of 2 to 4 years characterized by longer life expectancy, fertility, minimally reduced intellect and a better quality of life while severe forms present earlier with severe somatic manifestations, progressive cognitive impairment and death by the second decade of life due to cardiopulmonary complications.[12,13,14] The phenotypic expression of MPS II include coarse facial facies features, enlarged tongue, short stature, joint contractures skeletal abnormalities, cardiomyopathy, obstructive airway disease, retinopathy, cognitive deficits, hearing loss, sleep apnoea, visceromegaly and inguinal scrotal hernia. This phenotypic presentation should raise concern for the diagnosis and in resource-limited regions, is often the only resource available.[11]
Given the wide range of signs and symptoms for MPS II, confirmatory diagnosis is made by demonstrating a reduction/absence of IDS enzyme activity. A stepwise approach is recommended, with screening via urinary GAG levels (heparan and dermatan sulphatase), followed by confirmatory diagnosis using biochemical assay to check enzyme activity levels with dry blood spots and a molecular genetic analysis of the IDS gene.[13]
Enzyme replacement therapy for MPSII is now available and has shown great benefit in improving quality of life in these patients.[12,13] The management of this condition includes supportive care by a multidisciplinary team including pulmonology, cardiology, neurology, orthopaedics, otolaryngology, ophthalmology, and occupational and physical therapists. In most parts of Africa enzyme replacement therapy is not available. The care of these individuals is further compromised by the lack of specialists in most rural parts of Africa.
We report a second case of MPS II from Eastern Zambia, which highlights late diagnosis, as well as management strategies optimized via partnerships with higher specialized institutions in-country as well as potential more specialized treatment options through collaborations with international higher resourced centres.
Case presentation
An 11- year-old male (SP) was referred to Chipata Central Hospital (CCH) for concern of MPS II after recognition of concerning symptoms of fatigue and intellectual disability noted by a visiting Neuropsychologist to Zambia, who had been contacted by his local teachers. His older sibling who had since died had similar symptoms, which raised concern to this provider of a genetic condition. Through coordination between the University of Minnesota Neurodevelopmental Program in Rare Diseases, a neurological evaluation by a regularly visiting Child Neurologist to CCH (AAP) was arranged in coordination with a local paediatrics team. The child was accompanied by his home-based educator and mother.
SP was born to non-consanguineous parents as the second child of four. He had an unremarkable birth history. Early development was notable for global delay in the developmental milestones, head control was achieved around 8 months and ability to walk around 17 months, he spoke around 5 years of age and had challenges following through simple step instructions. Around the age of 2 years, child was noted to have lumpy swellings on the scalp and right side of the chest associated with easy fatigability. Since the age of 6 years, he has had recurrent diarrheal episodes, chest and ear infections. He started school at 7 years old with noted poor performance that he could not make progress with regular school and special education was recommended. He also did not make much progress in language after the age of 5 years, with his current comprehension and expression skills estimated by caregivers to be around the age of a 4- to 5-year-old.
During the last few years prior to presentation to CCH, he was noted to have worsening exertional fatigue, increased work of breathing, noisy breathing during sleep, headaches and concerns for progressive head enlargement and abdominal distention. He also become less talkative and more reserved, with increased fear for clinical settings in the background of his 15-year-old brother recently dying with a respiratory illness after a long-standing similar clinical picture.
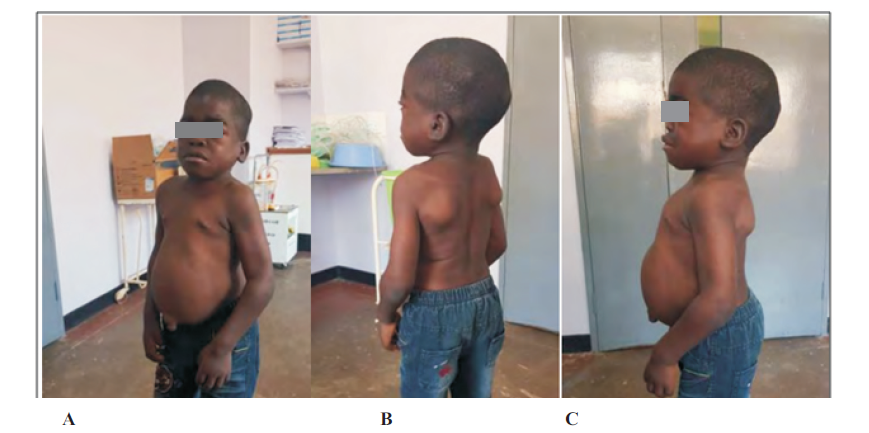
Physical exam revealed a child with short stature (Height for age below 5th percentile), macrocephaly (head circumference for age above 97th percentile) and multiple firm scalp mases. Dysmorphic features including coarse facial features, broad nasal bridge, large lips and tongue, wide set eyes, and slightly posterior rotated ears. There was no corneal clouding. He had visible mild increased work of breathing with increased anteroposterior chest diameter, mild tachycardia at rest with an appreciable diastolic heart murmur loudest at lower left sternal boarder. Abdomen was protruding and he had an umbilical hernia with hepatomegaly and splenomegaly at 10cm and 6cm below costal margin respectively. Spine had lumbar lordosis. (Figure 1).
He was very limited in his speech with the clinicians; however, he did speak in brief phrases in the local dialect with caregivers. Cranial nerve exam revealed appropriate pupillary reactions, extraocular movements, clear optic disc margin appreciable on left (right not appreciable, limited cooperation), intact visual fields, and symmetric facies. Hearing assessment was limited by significant bilateral wax impaction present. Tone was mildly increased throughout. He had grossly full and symmetric strength in all extremities. No dysmetria was noted and he displayed a hemiparetic gait, with right leg held stiffer and right shoulder shifting down while walking.
After identification of this family and coordination with a private laboratory and clinic and support from the US-based clinicians his siblings all had urine IDS enzyme assays two years prior to initial clinical evaluation at CCH. His recently deceased 15-year-old brother had obtained the testing soon before his death and was found to have results consistent with MPS II. SP’s younger brother and sister had normal enzyme activity. The three maternal uncles were all healthy.
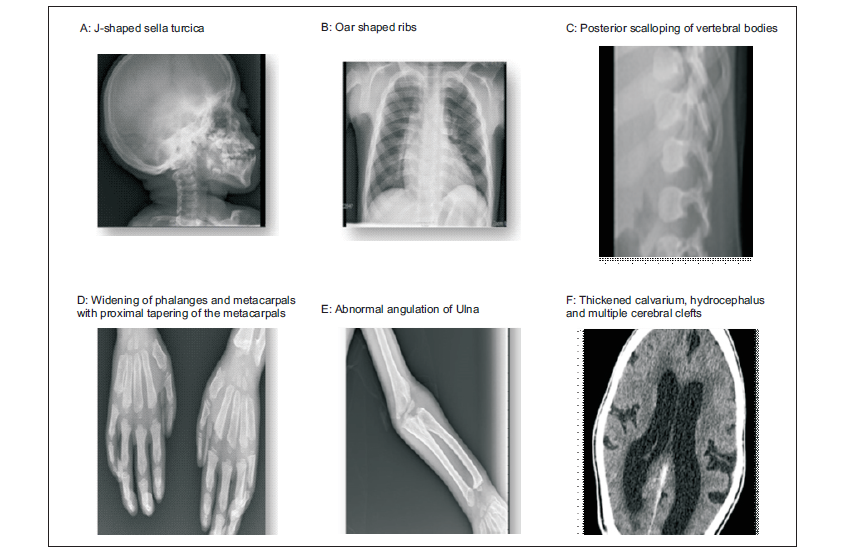
His full blood count, renal and liver function tests were unremarkable. An enzyme assay for IDS enzyme activity was at 0.0µmol/L/h (normal >2.5µmol/L/h). The enzyme activity for MPS I, IVA, VI and VII were normal. Mutational analysis was not done. Echocardiography showed thickened mitral valve with stenosis, thick aortic valve with aortic regurgitation with a normal ejection fraction. Radiography (Figure 2) showed thickened calvarium and j –shaped Sella turcica, oar-like ribs, posterior scalloping of vertebral bodies, ulna with abnormal angulation, widening of phalanges and metacarpals with proximal tapering of the metacarpals. A CT scan of the brain (Figure 2) revealed thickened calvarium, communicating hydrocephalus and multiple cerebral clefts.
Based upon his diagnostics and clinical presentation, coordination of care was arranged at University Teaching Hospital – Children’s Hospital, in Lusaka, the capital city of Zambia and approximately 570 kilometres away from the patient’s location. As the family is from a rural and lower socioeconomic background, trips to Lusaka required external supports and local care coordination. Our US-based partners continue to work through charitable organizations to seek out options for delivering enzyme replacement therapy to SP. In the interim, multi-disciplinary care is being sort with local paediatric neurology team, speech, behaviour and occupational therapy, cardiology, and gastroenterology through the mechanisms above.
DISCUSSION
MPS II is a severe but treatable condition in regions where early identification and enzyme replacement therapy is available. In resource-limited settings, such children are often not recognized, and when they are, clinical care options are limited severely. However, when recognized, administration of available supportive care can lengthen survival with good recognition and education about the risks of respiratory illnesses. In the case we presented above, the family had stopped seeking out care initially due to repeatedly being told nothing could be done, a tragic scenario that led to lack of care seeking for the older child when ill and subsequent early mortality.
Our case highlights some of the challenges faced in low resourced settings. These included low index of suspicion from clinicians of such rare diseases such that by the time a diagnosis is suspected, the disease is at advanced stage limiting treatment options which might have improved the quality of life if started at opportune time. The lack of specialized care, limited exposure, and lack of local diagnostic algorithms in the rural parts of Zambia, as in resource poor countries, poses a great challenge in early detection of such rare conditions. Furthermore, there is no direct capacity to conduct confirmatory studies, much less offer enzyme replacement therapy as highlighted from a recent case report from Zambia and other parts of sub-Saharan Africa.[11,15] Late diagnosis and awareness of the importance of supportive management strategies contribute to increased morbidity and early mortality in these patients.
The definite diagnosis of MPS II in this case was made due to a great collaboration at different levels. There is need for an improved engagement process between countries and institutions with diagnostic and treatment capacity for this rare condition to facilitate opportune intervention and thus improve quality of life for these children as demonstrated in recent studies.[12]
As more cases of these rare conditions are published, there will be increased sensitization among the clinicians across Zambia and other resource limited regions, which will lead to early case recognition and thus prompt early referral to higher centres for investigation and multidisciplinary care.
A multidisciplinary approach is warranted in these children for early identification of complications and specialized care. This will improve the quality of life even though enzyme replacement therapy is not a feasible option at the moment in Zambia. The emerging e-health services provide an excellent opportunity to facilitate linkage of these patients and provide tele-consults to the clinicians on the ground overcoming the geographical barriers.[16]
CONCLUSION
MPS II is lysosomal storage disorder with an insidious onset and non-reversible progressive course. Early diagnosis and opportune commencement of therapy is important to improve quality of life. Improved sensitization of clinicians on this condition and collaboration with countries and institutions with diagnostic and treatment capacity is warranted.
Patient perspective
The family was initially devasted with the death of the first born and being told repeatedly that nothing could be done for their second born child who was presenting with similar symptoms. However, after the above collaboration the family now have a diagnosis of the condition and are grateful to the team. Some improvement in the quality of life of the child has been noted and the clinical team will continue with the scheduled reviews.
DECLARATIONS
Acknowledgement
The authors would like to acknowledge all the colleagues involved in the care of this patient, staff at Chipata Central Hospital and Kakumbi clinic as primary care providers, the Time and Tide foundation for the constant support to the family during the travels for reviews and stay in Lusaka, staff at the university teaching Hospitals-children’s Hospital and the visiting neuropsychiatrist for identifying and facilitation in making the definitive diagnosis for MPS II. Lastly many thanks to the caregivers for granting us consent to write this paper.
Conflict of interest
The authors have no conflict of interest to declare.
Authors contribution
D Tembo, S Nkhoma, K L Nkole, N Kawatu and A.A. Patel were directly involved in the evaluation and clinical care of the patient. D Tembo wrote the first draft and all authors reviewed, edited, and approved the final version of this manuscript.
REFERENCES
- Sousa Martins R, Rocha S, Guimas A, Ribeiro R. Hunter Syndrome: The Phenotype of a Rare Storage Disease. Cureus. 2022;14(2):e21985.
- Gupta A, Uttarilli A, Dalal A, Girisha KM. Hunter syndrome with late age of presentation: clinical description of a case and review of the literature. BMJ Case Rep. 2015;2015.
- Martin R, Beck M, Eng C, Giugliani R, Harmatz P, Munoz V, et al. Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome). Pediatrics. 2008;121(2):e377-86.
- Semyachkina AN, Voskoboeva EY, Zakharova EY, Nikolaeva EA, Kanivets IV, Kolotii AD, et al. Case report: a rare case of Hunter syndrome (type II mucopolysaccharidosis) in a girl. BMC Med Genet. 2019;20(1):66.
- Jurecka A, Krumina Z, Żuber Z, Różdżyńska-Świątkowska A, Kłoska A, Czartoryska B, et al. Mucopolysaccharidosis type II in females and response to enzyme replacement therapy. Am J Med Genet A. 2012;158a(2):450-4.
- Tuschl K, Gal A, Paschke E, Kircher S, Bodamer OA. Mucopolysaccharidosis type II in females: case report and review of literature. Pediatr Neurol. 2005;32(4):270-2.
- Chinawa J, Adimora G, Obu H, Tagbo B, Ujunwa F, Onubogu I. Clinical Presentation of Mucopolysaccharidosis Type II (Hunter's Syndrome). Ann Med Health Sci Res. 2012;2(1):87-90.
- Mutesa L, Muganga N, Lissens W, Boemer F, Schoos R, Pierquin G, et al. Molecular analysis in two siblings African patients with severe form of Hunter Syndrome: identification of a novel (p.Y54X) nonsense mutation. J Trop Pediatr. 2007;53(6):434-7.
- Seedat YK, Narrandes R, Maharaj IC. Mucopolysaccharidosis (Hunter's syndrome) in a black family. S Afr Med J. 1981;59(10):342-3.
- Peterson EM, van der Walt A. Mucopolysaccharidosis (Hunter's syndrome) in a black family. S Afr Med J. 1981;60(16):609.
- Nchimba L, Mpabalwani, E., Inambao, M., & Kawatu, N. Diagnosis of Hunter Syndrome (Mucopolysaccharidosis Type II) in a Resource Limited Setting A Case Report from Zambia. Zambia Medical Journal of Zambia, 48(3), 335 - 338. 2022.
- Grant N, Sohn YB, Ellinwood NM, Okenfuss E, Mendelsohn BA, Lynch LE, et al. Timing is everything: Clinical courses of Hunter syndrome associated with age at initiation of therapy in a sibling pair. Mol Genet Metab Rep. 2022;30:100845.
- Semyachkina AN, Voskoboeva EY, Nikolaeva EA, Zakharova EY. Analysis of long-term observations of the large group of Russian patients with Hunter syndrome (mucopolysaccharidosis type II). BMC Med Genomics. 2021;14(1):71.
- Wraith JE, Scarpa M, Beck M, Bodamer OA, De Meirleir L, Guffon N, et al. Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur J Pediatr. 2008;167(3):267-77.
- Rasheeedah I, Patrick O, Abdullateef A, Mohammed A, Sherifat K, Gbadebo I. Challenges in the Management of Mucopolysaccharidosis Type II (Hunter's Syndrome) in a Developing Country: a Case Report. Ethiop J Health Sci. 2015;25(3):279-82.
- Giacalone A, Marin L, Febbi M, Franchi T, Tovani-Palone MR. eHealth, telehealth, and telemedicine in the management of the COVID-19 pandemic and beyond: Lessons learned and future perspectives. World J Clin Cases. 2022;10(8):2363-8.
Medical Journal of Zambia, Vol 51, 2
The Medical Journal of Zambia, ISSN 0047-651X, is published by the Zambia Medical Association.
© This is an Open Access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.